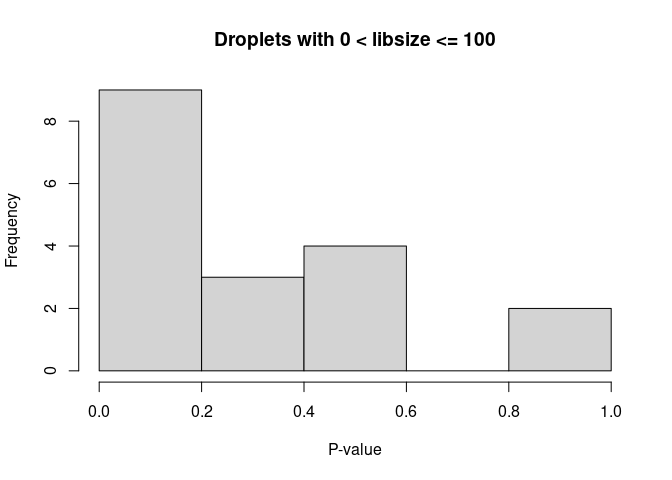
Visualize barcode rank plots for each sample
test <- demultiplexed_empty_joined %>% filter(demultiplexed_empty_joined$FDR < 0.001) %>% select(FDR, empty_droplet)
## tidyseurat says: Key columns are missing. A data frame is returned for independent data analysis.
test |> select(empty_droplet) |> table()
## empty_droplet
## FALSE
## 81090
Number of non-empty droplets, everything above knee is retained.
## Mode FALSE TRUE NA's
## logical 8429 81090 9215
## is.cell
## FALSE TRUE
## 8429 81090
Check if p-values are lower-bounded by ‘niters’ (increase ‘niters’ if any Limited==TRUE and Sig==FALSE), with niters being: An integer scalar specifying the number of iterations to use for the Monte Carlo p-value calculations.
## Limited
## Sig FALSE TRUE
## FALSE 8429 0
## TRUE 22814 58276
MA plot
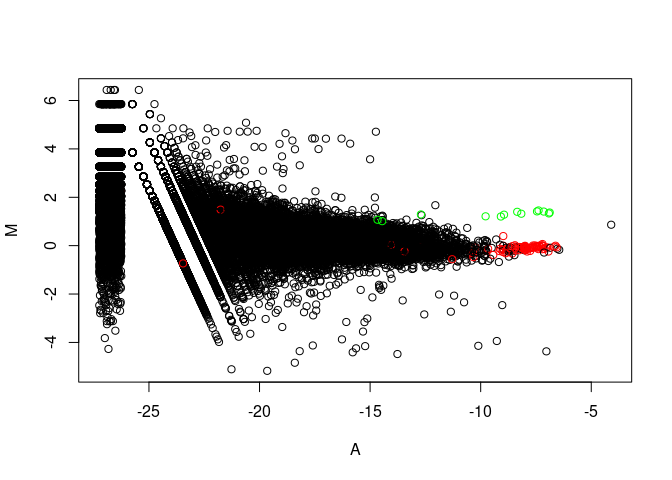
(mitochondria genes in green, ribosomal genes in red)
Histogram of p-values: